
Test de Compatibilidad Genética y Diagnóstico de enfermedades futuras. Avance de tecnología a pasos agigantados. Por Dra. Carmen Navarro y Dra. Lucy Coleman
 Siempre hemos insistido en que la tecnología va creciendo y nos aporta cada vez mas ayuda en el ámbito de la medicina innovadora. Contamos actualmente con el Test de Compatibilidad Genética (CGT) que es una prueba genética de importancia sobre todo en la planificación de la familia, ya que permite determinar el riesgo de tener un hijo con una enfermedad genética. El test informa si los progenitores son portadores de una o más mutaciones genéticas recesivas.
Siempre hemos insistido en que la tecnología va creciendo y nos aporta cada vez mas ayuda en el ámbito de la medicina innovadora. Contamos actualmente con el Test de Compatibilidad Genética (CGT) que es una prueba genética de importancia sobre todo en la planificación de la familia, ya que permite determinar el riesgo de tener un hijo con una enfermedad genética. El test informa si los progenitores son portadores de una o más mutaciones genéticas recesivas.
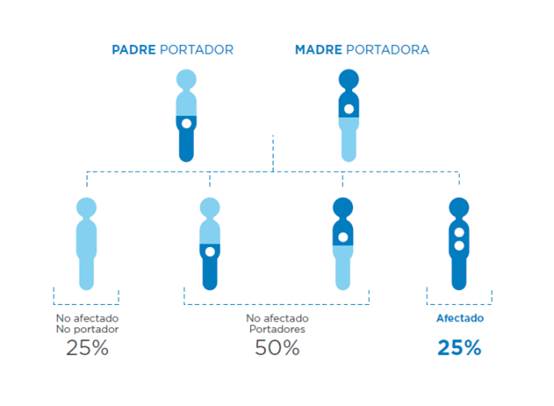
Los portadores suelen ser personas aparentemente sanas, pero cuando los dos padres son portadores de una mutación en el mismo gen, pueden dar lugar a un hijo afectado.
Cualquier persona puede ser, sin saberlo, portadora de una o más mutaciones.
El Test de Compatibilidad Genética nos permite saber qué genes tiene alterados cada persona.
¿Por qué hacerse un Test de Compatibilidad Genética?
Las enfermedades genéticas no se pueden curar, pero si se pueden prevenir. Generalmente los progenitores que son portadores de enfermedades genéticas graves se dan cuenta de ello después de dar a luz a un niño afectado. Este es un test válido clínicamente y es herramienta esencial para la detección de este tipo de enfermedades genéticas que pueden ocasionar daños mayores a futuro.
¿A quien va dirigido este test?
Todos tenemos alteraciones en nuestros genes y con este test podemos saber si éstas podrían causar una enfermedad a nosotros o a nuestros hijos. Se recomienda hacer el test en los siguientes casos:

– En caso de que usted desee saber cuál es su probabilidad de padecer alguna enfermedad. Lo cual le brinda una enorme ventaja, porque podría prevenirla o apoyar a grupos que se encuentran buscando la cura para su enfermedad, ej: Parkinson, Cáncer de mama.
– Antes de intentar por medios naturales un embarazo. Cualquier mujer que quiera embarazarse debería conocer los riesgos de transmitir posibles enfermedades a su hijo.
– Antes de un tratamiento de reproducción asistida. Es aconsejable para poder conocer el riesgo de transmisión y poder determinar el mejor tipo de tratamiento en cada caso.
– Antes de un tratamiento con óvulos o semen de donante. Para poder seleccionar un donante que no sea portador de la misma mutación que el miembro de la pareja que aporta los gametos (óvulos o espermatozoides).
¿Qué es una mutación genética?
Cada uno de los genes tiene dos copias, una heredada del padre y otra de la madre. Ser portador de una mutación genética significa que la persona tiene una alteración en una de las copias de un gen concreto.
¿Qué son los genes?
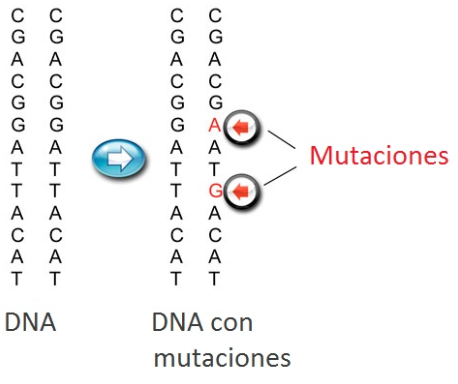
Cada una de nuestras células contiene información geneetica o ADN, organizada en unidades básicas, los genes, Los que no funcionan correctamente son responsables de enfermedades genéticas.
Los cambios en los genes se denominan mutaciones. Todos somos portadores de un número variable de las mismas.
Aunque los portadores son personas sanas, si ambos progenitores tienen una mutación en el mismo gen, la probabilidad de tener un hijo enfermo es del 25%.
¿Qué enfermedades se incluyen?
El Test de Compatibilidad Genética cubre un amplio rango de mutaciones que derivan en graves enfermedades genéticas. Incluye el cribado de todas las mutaciones recomendadas por los colegios profesionales de ginecología y genética.
Según datos de la Organización Mundial de la Salud (OMS), la prevalencia global de estas enfermedades es de 10 por cada 1000 recién nacidos.
Existen estimaciones que, tomadas en su conjunto, indican que estas enfermedades representan el 20% de las causas de mortalidad infantil en países desarrollados y están detrás del 18% de las actuaciones pediátricas hospitalarias. Las principales enfermedades monogénicas detectadas en este test son:
Fibrosis quística
Atrofia muscular espinal
Poliquistosis renal autosómica recesiva
Sordera hereditaria no sindrómica
Mucopolisacaridosis
Anemia falciforme
Enfermedad de Gaucher
Síndrome de X frágil
ß-Talasemia
Resultados
Cuando el resultado es positivo significa que la persona que se ha realizado el test es portadora de una mutación en un gen concreto. En este caso, el test se deberá llevar a cabo también en el otro integrante de la pareja. Si los dos son portadores de una mutación en el mismo gen, existe riesgo de tener hijos enfermos.
Un resultado negativo indica que la persona que se ha realizado el test no es portadora de las mutaciones estudiadas. Sin embargo, queda un riesgo residual de que la persona que se ha realizado el test aún sea portadora de otras mutaciones menos frecuentes no analizadas.
¿Qué hacer cuando ambos progenitores son positivos?
Si los dos miembros de la pareja obtienen un resultado positivo en el test con mutación para el mismo gen, la recomendación es consultar con el especialista acerca de las opciones para concebir un hijo sano.
Estas parejas pueden optar por un Diagnóstico Genético Pre-implantación (PGD) y evitar que su hijo sufra la enfermedad.

Otros pueden recurrir a la donación de gametos para evitar la transmisión de estas enfermedades.
Los padres pueden valorar la adopción para evitar tener in hijo enfermo.
Para realizar el test:
Solo debes acudir a nuestra unidad de fertilidad y se realiza una toma de sangre. Se coloca en un kit especial y se envía la muestra para su procesamiento. Los resultados se obtienen en aproximadamente 30-40 días.
Estudio de 15.000 mutaciones asociadas a más de 600 enfermedades genéticas.
Este test incluye también una secuenciación completa de mas de 549 genes correspondientes a más de 600 enfermedades genéticas.
CGT Match: este software permite seleccionar el gameto genéticamente compatible para un receptor. Este sistema permite a la clínica disponer de un banco genéticamente testado para las más de 600 enfermedades analizadas por el CGT. Incluye fibrosis quística y el síndrome de X-frágil.
Se realiza el CGT match al paciente que aporta los gametos. Una vez testados los donantes y la persona que aporta el gameto propio, nuestro programa CGT Match realizará la búsqueda para asignar un/una donante compatible, cuyas mutaciones no coincidan con las del paciente.
Su limitación pudiera ser su costo, sin embargo las ventajas son enormes.
 Nuestro listado de enfermedades detectadas por el CGT:
Nuestro listado de enfermedades detectadas por el CGT:
Abetalipoproteinemia
Acidemia glutárica tipo 2 (gen EFTFDH)
Acidemia glutárica tipo 2 (gen ETFA)
Acidemia glutárica tipo 2 (gen ETFB)
Acidemia isovalérica
Acidemia metilmalónica con homocistinuria tipo cblC
Acidemia metilmalónica con homocistinuria tipo cblD
Acidemia metilmalónica por déficit de metilmalonil-CoA epimerasa
Acidemia metilmalónica resistente a vitamina B12 tipo mut-
Acidemia metilmalónica vitamina B12 sensible tipo cbl A
Acidemia metilmalónica vitamina B12 sensible tipo cbl B
Acidemia propiónica (gen PCCA)
Acidemia propiónica (gen PCCB)
Acidosis láctica infantil fatal con aciduria metilmalónica
Aciduria 3-hidroxi-3-metil-glutárica
Aciduria 3-metilglutacónica tipo 1
Aciduria 3-metilglutacónica tipo 3
Aciduria 4 hidroxi-butírica
Aciduria argininosuccínica
Aciduria D-2-hidroxiglutárica
Aciduria fumárica
Aciduria malónica
Aciduria mevalónica
Acondrogénesis tipo 1B
Adrenoleucodistrofia neonatal (gen PEX12)
Adrenoleucodistrofia neonatal (gen PEX2)
Adrenoleucodistrofia neonatal (gen PEX26)
Adrenoleucodistrofia neonatal (gen PEX5)
Agammaglobulinemia ligada a X
Agenesia de cuerpo calloso – neuropatía
Agenesia de cuerpo calloso ligada al X con mutación en el gen Alfa 4
Agenesia pancreática
AICA ribosiduria
Albinismo ocular recesivo ligado al X
Albinismo oculocutáneo tipo 1A
Albinismo oculocutáneo tipo 2
Albinismo oculocutáneo tipo 3
Albinismo oculocutáneo tipo 4
Alcaptonuria
Alfa talasemia
Alfa-manosidosis
Amaurosis congénita de Leber
Amaurosis congénita de Leber
Amaurosis congénita de Leber
Amaurosis congénita de Leber 14
Amaurosis congénita de Leber 15
Amaurosis congénita de Leber 16
Amaurosis congénita de Leber 7
Amaurosis congénita de Leber 9
Anemia de Fanconi grupo de complementación A
Anemia de Fanconi grupo de complementación C
Anemia de Fanconi grupo de complementación D1
Anemia de Fanconi grupo de complementación D2
Anemia de Fanconi grupo de complementación E
Anemia de Fanconi grupo de complementación I
Anemia de Fanconi grupo de complementación J
Anemia de Fanconi grupo de complementación L
Anemia de Fanconi grupo de complementación M
Anemia de Fanconi grupo de complementación N
Anemia de Fanconi grupo de complementación O
Anemia de Fanconi grupo de complementación P
Anemia de Fanconi, grupo de complementación G
Anemia falciforme
Anemia hemolítica por déficit de piruvato quinasa de los glóbulos rojos
Anemia sideroblástica ligada al cromosoma X
Anemia sideroblástica ligada al X con ataxia
Aniridia – ataxia cerebelosa – déficit intelectual
Anoftalmia – microftalmia aisladas
Aplasia/hipoplasia de extremidades y pelvis
Argininemia
Arteriopatía cerebral autosómica recesiva con infarto subcortical y leucoencefalopatía (CARASIL)
Artrogriposis – disfunción renal – colestasis
Aspartilglucosaminuria
Ataxia – apraxia oculomotora tipo 1
Ataxia autosómica recesiva por déficit de ubiquinona
Ataxia con déficit de vitamina E
Ataxia de Friedreich
Ataxia espástica autosómica recesiva de Charlevoix-Saguenay
Ataxia espinocerebelosa con neuropatía axonal tipo 2
Ataxia espinocerebelosa infantil
Ataxia letal con sordera y atrofia óptica
Atelosteogénesis tipo 2
Atrofia espinal distal autosómica recesiva tipo 4
Atrofia girada de la coroides y la retina
Atrofia muscular espinal con insuficiencia respiratoria
Atrofia muscular espinal distal ligada al X
Atrofia muscular espinal ligada al X tipo 2
Atrofia muscular espinal proximal tipo 1
Atrofia muscular espinal proximal tipo 2
Atrofia muscular espinal proximal tipo 3
Atrofia muscular espinal proximal tipo 4
Beta-talasemia
Catarata – déficit intelectual – hipogonadismo
Catarata congénita – dismorfismo facial – neuropatía
Ceguera nocturna estacionaria congénita
Ceguera nocturna estacionaria congénita
Ceguera nocturna estacionaria congénita tipo 1B
Ceguera nocturna estacionaria congénita tipo 1E
Cetoacidosis por déficit de beta-cetotiolasa
Cistinosis
Citrulinemia tipo 2 forma adulta
Citrulinemia tipo 2 forma neonatal
Citrulinemia tipo I
Complejo xeroderma pigmentoso/síndrome de Cockayne, grupo de complementación B
Complejo xeroderma pigmentoso/síndrome de Cockayne, grupo de complementación D
Complejo xeroderma pigmentoso/síndrome de Cockayne, grupo de complementación F
Complejo xeroderma pigmentoso/síndrome de Cockayne, grupo de complementación G
Condrodisplasia punctata braquitelefalángica
Condrodisplasia punctata rizomélica tipo 1
Condrodisplasia punctata rizomélica tipo 3
Convulsiones sensibles al piridoxal fosfato
Coreoacantocitosis
Defecto congénito en la síntesis de ácidos biliares tipo 3
Deficiencia de piruvato deshidrogenasa fosfatasa
Déficit aislado de CoQ-citocromo C reductasa
Déficit aislado de hormona de crecimiento tipo III
Déficit aislado de la hormona estimulante de la tiroides
Déficit combinado de la fosforilación oxidativa tipo 2
Déficit combinado de la fosforilación oxidativa tipo 5
Déficit congénito de fibrinógeno (gen FGA)
Déficit congénito de fibrinógeno (gen FGB)
Déficit congénito de síntesis de ácidos biliares tipo 4
Déficit congénito del factor V
Déficit congénito del factor XI
Déficit de 2-metilbutiril-CoA deshidrogenasa
Déficit de 3-hidroxiacil-CoA deshidrogenasa de ácidos grasos de cadena larga
Déficit de 3-metilcrotonil-CoA carboxilasa tipo 1
Déficit de 3-metilcrotonil-CoA carboxilasa tipo 2
Déficit de acil CoA oxidasa peroxisomal
Déficit de acil-CoA deshidrogenasa 9
Déficit de acil-CoA deshidrogenasa de cadena corta
Déficit de acil-CoA deshidrogenasa de cadena media
Déficit de acil-CoA deshidrogenasa de cadena muy larga
Déficit de alfa-1 antitripsina
Déficit de Alfa-metilacil-CoA racemasa
Déficit de alfa-N-acetilgalactosaminidasa tipo 1
Déficit de alfa-N-acetilgalactosaminidasa tipo 2
Déficit de biotinidasa
Déficit de carbamil-fosfato sintetasa
Déficit de carnitina palmitoiltransferasa 1A
Déficit de carnitina palmitoiltransferasa II, forma infantil
Déficit de carnitina palmitoiltransferasa II, forma neonatal
Déficit de dihidropirimidina deshidrogenasa
Déficit de enzima bifuncional
Déficit de fosfoserina aminotransferasa
Déficit de glutaril-CoA deshidrogenasa
Déficit de glutatión sintetasa con 5-oxoprolinuria
Déficit de guanidinoacetato metiltransferasa
Déficit de hormonas hipofisarias combinado con anomalías de la columna vertebral
Déficit de L-aminoácido aromático decarboxilasa
Déficit de metionina adenosiltransferasa
Déficit de ornitina transcarbamilasa
Déficit de piruvato carboxilasa
Déficit de plasminógeno tipo 1
Déficit de proteina trifuncional mitocondrial
Déficit de proteina trifuncional mitocondrial
Déficit de sulfito oxidasa por déficit del cofactor molibdeno tipo A (gen MOCS1)
Déficit de sulfito oxidasa por déficit del cofactor molibdeno tipo A (gen MOCS2)
Déficit de transportador de creatina ligado al X
Déficit del complejo 3 de la cadena respiratoria mitocondrial
Déficit del complejo 3 de la cadena respiratoria mitocondrial
Déficit del componente 3 del complemento
Déficits combinados de hormonas hipofisarias de causas genéticas
Déficits combinados de hormonas hipofisarias de causas genéticas
Déficits combinados de hormonas hipofisarias de causas genéticas
Dermopatía restrictiva letal
Dermopatía restrictiva letal
Diarrea congénita con malabsoción por insuficiencia de células enteroendocrinas
Disautonomía familiar
Disgenesia tubular renal
Disgenesia tubular renal
Disgenesia tubular renal
Disgenesia tubular renal
Disostosis espondilocostal autosómica recesiva 1
Displasia de Greenberg
Displasia de Schneckenbecken
Displasia ectodérmica – ectrodactilia – distrofia macular
Displasia ectodérmica hipohidrótica
Displasia epifisaria múltiple tipo 4
Displasia espondiloepimetafisaria tipo matrilina-3
Displasia frontometafisaria
Displasia geleofísica 1
Displasia letal osteosclerótica de hueso
Displasia mandíbuloacra con lipodistrofia tipo A
Displasia mandíbuloacra con lipodistrofia tipo B
Displasia oculodentodigital
Displasia odonto-ónico-dérmica
Displasia oto-espondilo-megaepifisaria
Displasia renal-hepática-pancreática
Disqueratosis congénita 2
Disqueratosis congénita autosomica recesiva 2
Disqueratosis congénita autosomica recesiva 3
Disqueratosis congénita ligada al X
Distonía 16
Distonia dopa-sensible autosómica recesiva
Distonía-parkinsonismo ligado al X
Distrofia corneal macular 1
Distrofia cristalina de Bietti
Distrofia de cintura con epidermólisis bullosa simple
Distrofia de conos con respuesta escotópica supranormal
Distrofia de conos progresiva 4
Distrofia de conos y bastones 10
Distrofia de conos y bastones 15
Distrofia de conos y bastones 3
Distrofia de córnea – sordera de percepción
Distrofia de retina de Bothnia
Distrofia endotelial hereditaria congénita II
Distrofia muscular congénita asociada a anomalías cerebrales
Distrofia muscular congénita asociada a anomalías cerebrales
Distrofia muscular congénita asociada a anomalías cerebrales
Distrofia muscular congénita tipo 1A
Distrofia muscular congénita tipo 1D
Distrofia muscular congénita tipo 4B
Distrofia muscular congénita tipo 5B
Distrofia muscular congénita tipo Fukuyama
Distrofia muscular de cinturas autosómica recesiva tipo 2A
Distrofia muscular de cinturas autosómica recesiva tipo 2B
Distrofia muscular de cinturas autosómica recesiva tipo 2C
Distrofia muscular de cinturas autosómica recesiva tipo 2D
Distrofia muscular de cinturas autosómica recesiva tipo 2E
Distrofia muscular de cinturas autosómica recesiva tipo 2G
Distrofia muscular de cinturas autosómica recesiva tipo 2H
Distrofia muscular de cinturas autosómica recesiva tipo 2J
Distrofia muscular de cinturas autosómica recesiva tipo 2l
Distrofia muscular de cinturas autosómica recesiva tipo 2L
Distrofia muscular de cinturas autosómica recesiva tipo 2M
Distrofia muscular de cinturas autosómica recesiva tipo C
Distrofia muscular de cinturas autosómica recesiva tipo C
Distrofia muscular de cinturas autosómica recesiva tipo C
Distrofia muscular de Duchenne
Distrofia muscular de Emery-Dreifuss
Distrofia muscular de Emery-Dreifuss 6
Distrofia muscular tipo Becker
Distrofia neuroaxonal infantil 2A
Distrofia neuroaxonal infantil 2B
Enanismo diastrófico
Enanismo MULIBREY
Encefalomiopatía neurogastrointestinal mitocondrial
Encefalopatía debida a una deficiencia de prosaposina
Encefalopatía epiléptica infantil temprana
Encefalopatía epiléptica infantil temprana
Encefalopatía etilmalónica
Encefalopatía grave de aparición neonatal con microcefalia
Enfermedad congénita de glucosilación tipo 1t
Enfermedad de almacenamiento de ácido siálico libre, forma infantil
Enfermedad de almacenamiento de glucógeno por déficit de aldolasa A
Enfermedad de almacenamiento de glucógeno por déficit de beta-enolasa muscular
Enfermedad de almacenamiento de glucógeno por déficit de enzima desramificante del glucógeno
Enfermedad de almacenamiento de glucógeno por déficit de enzima desramificante, forma combinada hepática y miopática infantil
Enfermedad de almacenamiento de glucógeno por déficit de fosfoglucomutasa
Enfermedad de almacenamiento de glucógeno por déficit de glucógeno fosforilasa muscular
Enfermedad de almacenamiento de glucógeno por déficit de glucosa-6-fosfatasa tipo b
Enfermedad de almacenamiento de glucógeno por déficit de glucosa-6-fosfatasa tipo c
Enfermedad de almacenamiento de glucógeno por déficit de la subunidad M de la lactato deshidrogenasa
Enfermedad de almacenamiento de glucógeno por déficit de maltasa ácida
Enfermedad de almacenamiento de glucógeno tipo 9C
Enfermedad de astas posteriores, ataxia – Retinosis pigmentaria
Enfermedad de Canavan
Enfermedad de Charcot-Marie-Tooth autosómica dominante tipo 2K
Enfermedad de Charcot-Marie-Tooth autosómica recesiva con ronquera
Enfermedad de Charcot-Marie-Tooth intermedia autosómica recesiva tipo A
Enfermedad de Charcot-Marie-Tooth ligada al X tipo 5
Enfermedad de Charcot-Marie-Tooth tipo 1F
Enfermedad de Charcot-Marie-Tooth tipo 2B2
Enfermedad de Charcot-Marie-Tooth tipo 4A
Enfermedad de Charcot-Marie-Tooth tipo 4B1
Enfermedad de Charcot-Marie-Tooth tipo 4B2
Enfermedad de Charcot-Marie-Tooth tipo 4C
Enfermedad de Charcot-Marie-Tooth tipo 4D
Enfermedad de Charcot-Marie-Tooth tipo 4E
Enfermedad de Charcot-Marie-Tooth tipo 4E
Enfermedad de Charcot-Marie-Tooth tipo 4F
Enfermedad de Charcot-Marie-Tooth tipo 4H
Enfermedad de Charcot-Marie-Tooth tipo 4J
Enfermedad de Charcot-Marie-Tooth tipo axonal 2B1
Enfermedad de Dent
Enfermedad de Gaucher forma fetal
Enfermedad de Gaucher tipo 2
Enfermedad de Gaucher tipo 3
Enfermedad de Gaucher tipo 3C
Enfermedad de Griscelli tipo 1
Enfermedad de Griscelli tipo 2
Enfermedad de jarabe de arce clásica
Enfermedad de Kennedy
Enfermedad de Krabbe
Enfermedad de Krabbe
Enfermedad de la orina de jarabe de arce
Enfermedad de la orina de jarabe de arce (gen BCKDHA)
Enfermedad de la orina de jarabe de arce (gen BCKDHB)
Enfermedad de Menkes
Enfermedad de músculo-ojo-cerebro
Enfermedad de músculo-ojo-cerebro
Enfermedad de Niemann-Pick tipo A
Enfermedad de Niemann-Pick tipo B
Enfermedad de Niemann-Pick tipo C1
Enfermedad de Niemann-Pick tipo C2
Enfermedad de Oguchi
Enfermedad de Refsum
Enfermedad de Sandhoff
Enfermedad de Stargardt
Enfermedad de Stargardt 1
Enfermedad de Stargardt 1
Enfermedad de Tay-Sachs
Enfermedad de Thomsen y Becker
Enfermedad de Unverricht-Lundborg
Enfermedad de Wilson
Enfermedad linfoproliferativa ligada al X
Enfermedad mitocondrial fatal debida a una deficiencia de fosforilación oxidativa tipo 3 combinada
Enfermedad tipo Pelizaeus-Merzbacher debida a mutación en GJC2
Enfermedad tipo Pelizaeus-Merzbacher debida a mutación en HSPD1
Epidermólisis bullosa acantolítica letal
Epidermólisis bullosa distrófica generalizada grave
Epidermólisis bullosa distrófica pruriginosa
Epidermólisis bullosa juntural – atresia pilórica
Epidermólisis bullosa juntural con atresia pilórica
Epidermólisis bullosa juntural generalizada tipo no Herlitz
Epidermólisis bullosa juntural tipo Herlitz (gen LAMA3)
Epidermólisis bullosa juntural tipo Herlitz (gen LAMB3)
Epidermólisis bullosa juntural tipo Herlitz (gen LAMC2)
Epidermólisis bullosa juntural tipo no Herlitz
Epidermólisis bullosa juntural tipo no Herlitz (gen LAMA3)
Epidermólisis bullosa juntural tipo no Herlitz (gen LAMB3)
Epidermólisis bullosa juntural tipo no Herlitz (gen LAMC2)
Epidermólisis bullosa simple con atresia pilórica
Epidermólisis bullosa simple con distrofia muscular
Epilepsia progresiva – déficit intelectual, tipo finlandés
Fenilcetonuria
Fibrocondrogenesis 1
Fibrosis quística; mucoviscidosis
Fiebre mediterránea familiar
Fucosidosis
Galactosemia clásica
Gangliosidosis GM1 tipo 1
Gangliosidosis GM1 tipo 2
Gangliosidosis GM1 tipo 3
Gangliosidosis GM2, variante AB
Glomeruloesclerosis focal segmental
Glucogenosis por déficit de glucosa-6-fosfatasa tipo 1a
Hábito marfanoide con déficit intelectual ligado al X
Hemocromatosis tipo 1
Hemocromatosis tipo 3
Hemofilia A
Hemofilia B
Hepatoencefalopatía por déficit combinado de la fosforilación oxidativa tipo 1
Hiperamoniaquemia por déficit de N-Acetilglutamato sintasa
Hiperglicinemia no cetósica aislada
Hiperglicinemia no cetósica aislada
Hiperglicinemia no cetósica aislada
Hiperornitinemia – hiperamonemia – homocitrulinuria
Hiperoxaluria primaria tipo 1
Hiperoxaluria primaria tipo 2
Hiperplasia suprarrenal congénita clásica por déficit de 21-hidroxilasa
Hiperplasia suprarrenal congénita lipoide
Hiperprolinemia tipo 1
Hiperprolinemia tipo 2
Hipofosfatasia de la infancia
Hipofosfatasia infantil
Hipomagnesemia familiar – hipercalciuria – nefrocalcinosis – afectación ocular grave
Hipomielinización – catarata congénita
Hipoparatiroidismo – déficit intelectual – dismorfismo
Hipoplasia adrenal congénita citomegálica
Hipoplasia o aplasia fibular – inclinación femoral – oligodactilia
Hipoplasia pontocerebelosa tipo 2
Hipoplasia pontocerebelosa tipo 4
Hipotiroidismo debido a mutaciones en el receptor TSH
Histidinemia
Homocistinuria clásica
Homocistinuria por déficit de metilentetrahidrofolato reductasa
Ictiosis folicular – alopecia – fotofobia
Inmunodeficiencia combinada con granulomas en la piel
Inmunodeficiencia combinada con granulomas en la piel
Inmunodeficiencia combinada grave por déficit completo de RAG1/2
Inmunodeficiencia combinada grave por déficit completo de RAG1/2
Inmunodeficiencia combinada grave por déficit de adenosina desaminasa
Inmunodeficiencia combinada grave por déficit de DCLRE1C
Inmunodeficiencia combinada grave T-B+ ligada al X
Inmunodeficiencia combinada grave T-B+ por déficit de cadena gamma
Inmunodeficiencia combinada grave T-B+ por déficit de JAK3
Inmunodeficiencia con anomalía de factor H
Inmunodeficiencia severa combinada con sensibilidad a radiación ionizante
Inmunodeficiencia severa de linfocitos T – alopecia congénita – distrofia ungueal
Intolerancia hereditaria a la fructosa
Latosterolosis
Leprechaunismo
Leucodistrofia metacromática
Leucodistrofia metacromática
Leucoencefalopatía megalencefálica con quistes subcorticales
Lipodistrofia tipo Berardinelli
Lipofuscinosis neuronal ceroide 2
Lipofuscinosis neuronal ceroide del adulto
Lipofuscinosis neuronal ceroide del adulto 10
Lipofuscinosis neuronal ceroide del adulto 4A
Lipofuscinosis neuronal ceroide juvenil 3
Lipofuscinosis neuronal ceroide tardía infantil
Lipofuscinosis neuronal ceroide tardía infantil 5
Lipofuscinosis neuronal ceroide tardía infantil 6
Lipofuscinosis neuronal ceroide tardía infantil 8
Microcefalia primaria autosómica recesiva
Microcefalia primaria autosómica recesiva
Microcefalia primaria autosómica recesiva
Microcefalia primaria autosómica recesiva 1
Microcefalia primaria autosómica recesiva 2
Microcefalia primaria autosómica recesiva 6
Microcefalia primaria autosómica recesiva 9
Microftalmia – cataratas
Microftalmia – Retinosis pigmentaria – foveosquisis – drusen de disco óptico
Microftalmia sindrómica tipo 9
Miocardiopatía dilatada con ataxia
Miopatía centronuclear ligada al X
Miopatía con cuerpos de inclusión tipo 2
Miopatía congénita multicore con oftalmoplejia externa
Miopatía de Miyoshi
Miopatía de Miyoshi
Miopatía distal de inicio en músculo tibial anterior
Miopatía distal tipo Nonaka
Miopatía hereditaria con acidosis láctica por déficit de ISCU
Miopatía nemalínica
Miopatía nemalínica 2
Mucolipidosis tipo 2
Mucolipidosis tipo 3
Mucolipidosis tipo 4
Mucopolisacaridosis tipo 2
Mucopolisacaridosis tipo 3A (Síndrome de Sanfilippo tipo A)
Mucopolisacaridosis tipo 3D (Enfermedad de Sanfilippo)
Mucopolisacaridosis tipo 4B
Mucopolisacaridosis tipo 6
Mucopolisacaridosis tipo 7
Nanoftalmia
Necrosis estriatal bilateral infantil
Neurodegeneración asociada a pantotenato-quinasa
Neurodegeneración por déficit en 3-hidroxisobutiril-CoA-hidrolasa
Neurohepatopatía tipo Navajo
Neuropatía atáxica sensitiva – disartria – oftalmoplejía
Neuropatía axonal gigante
Neuropatía sensitiva y autonómica tipo 4
Neutropenia congénita grave ligada al X
Oftalmoplejía externa progresiva autosómica recesiva
Osteogénesis imperfecta tipo 7
Osteogénesis imperfecta tipo 8
Osteopetrosis con acidosis tubular renal
Osteopetrosis con displasia neuroaxonal, forma infantil
Osteopetrosis maligna autosómica recesiva 1
Osteopetrosis maligna autosómica recesiva 4
Osteoporosis – pseudoglioma
Paraplejía espástica autosómica recesiva tipo 11
Paraplejía espástica autosómica recesiva tipo 15
Paraplejía espástica autosómica recesiva tipo 20
Paraplejía espástica autosómica recesiva tipo 7
Paraplejía espástica ligada al X tipo 2
Picnodisostosis
Poliquistosis renal autosómica recesiva
Pseudohipoaldosteronismo autosómico recesivo tipo 1 (gen SCNN1A)
Pseudohipoaldosteronismo autosómico recesivo tipo 1 (gen SCNN1B)
Pseudohipoaldosteronismo autosómico recesivo tipo 1 (gen SCNN1G)
Raquitismo dependiente de vitamina D tipo 2A
Raquitismo hipofosfatémico autosómico recesivo 1
Raquitismo hipofosfatémico autosómico recesivo 2
Retinopatía tipo Burgess-Black
Retinosis pigmentaria 10
Retinosis pigmentaria 12
Retinosis pigmentaria 14
Retinosis pigmentaria 19
Retinosis pigmentaria 2
Retinosis pigmentaria 20
Retinosis pigmentaria 25
Retinosis pigmentaria 26
Retinosis pigmentaria 3
Retinosis pigmentaria 35
Retinosis pigmentaria 36
Retinosis pigmentaria 37
Retinosis pigmentaria 38
Retinosis pigmentaria 4
Retinosis pigmentaria 40
Retinosis pigmentaria 41
Retinosis pigmentaria 43
Retinosis pigmentaria 44
Retinosis pigmentaria 45
Retinosis pigmentaria 46
Retinosis pigmentaria 47
Retinosis pigmentaria 49
Retinosis pigmentaria 55
Retinosis pigmentaria 56
Retinosis pigmentaria 57
Retinosis pigmentaria 59
Retinosis pigmentaria 62
Retraso en el crecimiento por déficit en el factor de crecimiento insulínico tipo 1
Secuencia deformante de aquinesia fetal
Síndrome ABCD
Síndrome acrocalloso
Síndrome COACH
Síndrome COFS 1
Síndrome de Aicardi-Goutières
Síndrome de Alpers
Síndrome de Alport autosómico recesivo (gen COL4A3)
Síndrome de Alport autosómico recesivo (gen COL4A4)
Síndrome de Alström
Síndrome de ataxia cerebelosa – déficit intelectual – desequilibrio
Síndrome de Bartter con sordera
Síndrome de Bartter prenatal
Síndrome de Bartter prenatal tipo 1
Síndrome de Björnstadt
Síndrome de Carpenter
Síndrome de Cockayne tipo A
Síndrome de Cockayne tipo B
Síndrome de Cohen tipo 1
Síndrome de columna rígida
Síndrome de contracturas congénitas letales tipo 1
Síndrome de córnea frágil
Síndrome de Crigler-Najjar tipo 1
Síndrome de Crigler-Najjar tipo 2
Síndrome de depleción del ADN mitocondrial, forma hepatocerebral por déficit de DGUOK 3
Síndrome de depleción del ADN mitocondrial, forma miopática
Síndrome de disgenesia cerebral – neuropatía – ictiosis – queratodermia palmoplantar
Síndrome de Donnai-Barrow
Síndrome de Dursun
Síndrome de Ehlers-Danlos tipo 6
Síndrome de Ehlers-Danlos tipo 7C
Síndrome de Ehlers-Danlos tipo cardiaco valvular
Síndrome de Fraser (gen FRAS1)
Síndrome de Fraser (gen FREM2)
Síndrome de Gilbert
Síndrome de Goldmann-Favre
Síndrome de Hermansky-Pudlak
Síndrome de hiper-IgM ligado al X
Síndrome de hipoplasia del cuerpo calloso – retraso – pulgares en aducción – espasticidad – hidrocefalia
Síndrome de Hoyeraal-Hreidarsson
Síndrome de insensibilidad completa a los andrógenos
Síndrome de insensibilidad parcial a los andrógenos
Síndrome de Jeune
Síndrome de Johanson-Blizzard
Síndrome de Joubert
Síndrome de Joubert
Síndrome de Joubert
Síndrome de Joubert 4
Síndrome de Joubert con defecto hepático
Síndrome de Joubert con defecto hepático
Síndrome de Joubert con defecto ocular
Síndrome de Joubert con defecto óculo-renal
Síndrome de Joubert con defecto óculo-renal 5
Síndrome de Joubert con defecto óculo-renal, digénico
Síndrome de Kelley-Seegmiller
Síndrome de Knobloch 1
Síndrome de Leigh
Síndrome de Leigh
Síndrome de Leigh con síndrome nefrótico
Síndrome de Leigh con síndrome nefrótico
Síndrome de Leigh ligado al X
Síndrome de Leigh tipo franco-canadiense
Síndrome de Lesch-Nyhan
Síndrome de lisencefalia tipo Norman-Roberts
Síndrome de Marinesco-Sjogren
Síndrome de Masa
Síndrome de McKusick Kaufman
Síndrome de Meckel
Síndrome de Meckel
Síndrome de Meckel tipo 1
Síndrome de Meckel tipo 5
Síndrome de Mohr-Tranebjaerg
Síndrome de Omenn
Síndrome de Omenn (gen RAG1)
Síndrome de Omenn (gen RAG2)
Síndrome de Perrault
Síndrome de Roberts
Síndrome de Sanfilippo tipo C
Síndrome de Schopf-Schulz-Passarge
Síndrome de Schwartz-Jampel
Síndrome de Seckel
Síndrome de Seckel
Síndrome de Senior-Loken
Síndrome de Senior-Loken
Síndrome de Senior-Loken 1
Síndrome de Senior-Loken 5
Síndrome de Shwachman-Diamond
Síndrome de Simpson-Golabi-Behmel tipo 2
Síndrome de Smith-Lemli-Opitz
Síndrome de Stickler autosómico recesivo tipo 4
Síndrome de Stickler autosómico recesivo tipo 5
Síndrome de Stuve-Wiedemann
Síndrome de sudoración inducida por frío
Síndrome de Usher
Síndrome de Usher tipo 1
Síndrome de Usher tipo 1C
Síndrome de Usher tipo 1D
Síndrome de Usher tipo 1F
Síndrome de Usher tipo 1G
Síndrome de Usher tipo 2A
Síndrome de Usher tipo 2A
Síndrome de Usher tipo 2C
Síndrome de Usher tipo 2C
Síndrome de Usher tipo 3A
Síndrome de Waardenburg tipo 2D
Síndrome de Waardenburg tipo 3
Síndrome de Waardenburg-Shah 4A
Síndrome de Waardenburg-Shah 4B
Síndrome de Walker-Warburg (gen POMT1)
Síndrome de Walker-Warburg (gen POMGNT1)
Síndrome de Walker-Warburg (gen POMT2)
Síndrome de Wiskott-Aldrich
Síndrome de Wolcott-Rallison
Síndrome de Wolfram
Síndrome de X-frágil
Síndrome de Zellweger 1A
Síndrome de Zellweger 7A
Síndrome del cuerno occipital
Síndrome GRACILE
Síndrome hydrolethalus 1
Síndrome hydrolethalus 2
Síndrome Micro
Síndrome nefrótico congénito tipo finlandés
Síndrome nefrótico idiopático resistente a esteroides con hialinosis segmentaria focal, forma familiar
Síndrome nefrótico tipo 3
Síndrome óculo-cerebro-renal
Sordera – encefaloneuropatía – obesidad – valvulopatía
Sordera mixta ligada al X con gusher perilinfático
Sordera neurosensorial no sindrómica autosómica recesiva, tipo DFNB 35
Sordera neurosensorial no sindrómica autosómica recesiva, tipo DFNB 36
Sordera neurosensorial no sindrómica autosómica recesiva, tipo DFNB10
Sordera neurosensorial no sindrómica autosómica recesiva, tipo DFNB11
Sordera neurosensorial no sindrómica autosómica recesiva, tipo DFNB16
Sordera neurosensorial no sindrómica autosómica recesiva, tipo DFNB18
Sordera neurosensorial no sindrómica autosómica recesiva, tipo DFNB1A (gen GJB2), digénica
Sordera neurosensorial no sindrómica autosómica recesiva, tipo DFNB1A (gen GJB3), digénica
Sordera neurosensorial no sindrómica autosómica recesiva, tipo DFNB1B, digénica
Sordera neurosensorial no sindrómica autosómica recesiva, tipo DFNB2
Sordera neurosensorial no sindrómica autosómica recesiva, tipo DFNB21
Sordera neurosensorial no sindrómica autosómica recesiva, tipo DFNB212
Sordera neurosensorial no sindrómica autosómica recesiva, tipo DFNB22
Sordera neurosensorial no sindrómica autosómica recesiva, tipo DFNB23
Sordera neurosensorial no sindrómica autosómica recesiva, tipo DFNB24
Sordera neurosensorial no sindrómica autosómica recesiva, tipo DFNB25
Sordera neurosensorial no sindrómica autosómica recesiva, tipo DFNB28
Sordera neurosensorial no sindrómica autosómica recesiva, tipo DFNB29
Sordera neurosensorial no sindrómica autosómica recesiva, tipo DFNB3
Sordera neurosensorial no sindrómica autosómica recesiva, tipo DFNB30
Sordera neurosensorial no sindrómica autosómica recesiva, tipo DFNB31
Sordera neurosensorial no sindrómica autosómica recesiva, tipo DFNB37
Sordera neurosensorial no sindrómica autosómica recesiva, tipo DFNB39
Sordera neurosensorial no sindrómica autosómica recesiva, tipo DFNB4
Sordera neurosensorial no sindrómica autosómica recesiva, tipo DFNB49
Sordera neurosensorial no sindrómica autosómica recesiva, tipo DFNB59
Sordera neurosensorial no sindrómica autosómica recesiva, tipo DFNB6
Sordera neurosensorial no sindrómica autosómica recesiva, tipo DFNB61
Sordera neurosensorial no sindrómica autosómica recesiva, tipo DFNB63
Sordera neurosensorial no sindrómica autosómica recesiva, tipo DFNB67
Sordera neurosensorial no sindrómica autosómica recesiva, tipo DFNB77
Sordera neurosensorial no sindrómica autosómica recesiva, tipo DFNB79
Sordera neurosensorial no sindrómica autosómica recesiva, tipo DFNB9
Sulfocisteinuria
Tirosinemia tipo 1
Tirosinemia tipo 2
Tirosinemia tipo 3
Trastorno congénito de la glicosilación tipo 1a
Trastorno congénito de la glicosilación tipo 1b
Trastorno congénito de la glicosilación tipo 1c
Trastorno congénito de la glicosilación tipo 1k
Trastorno congénito de la glicosilación tipo 2a
Trastorno congénito de la glicosilación tipo 2c
Trastorno congénito de la glicosilación tipo 2d
Trastorno congénito de la glicosilación tipo 2f
Trastorno congénito de la glicosilación tipo Ie
Trastorno congénito de la glicosilación tipo Ij
Vitreorretinopatía exudativa familiar 4
Xeroderma pigmentoso grupo de complementación A
Xeroderma pigmentoso grupo de complementación E
Para mayor información pueden contactarnos a través de nuestro email: [email protected] ó nuestro Nro. Master 0212-9753060.